

Knochenkrebs bei Kindern und Jugendlichen: Das Ewing-Sarkom
Primäre bösartige Knochentumore, sogenannte Sarkome, stellen eine seltene Krebserkrankung dar. Bestimmte aggressive Tumore dieser Krebsart, die sogenannten Ewing-Sarkome, treten hauptsächlich bei Kindern und Jugendlichen auf. Die Symptome äußern sich oft unspezifisch, was eine Diagnose erschwert. Werden mittels bildgebender Verfahren und einer Biopsie ein Ewing-Sarkom und möglicherweise auch Metastasen festgestellt, ist eine schnelle Therapie nach neuesten Standards überlebenswichtig. Heutige Behandlungsverfahren ermöglichen verbesserte Prognosen und Heilungschancen.
Inhaltsverzeichnis
Ein kurzer Überblick
Die wichtigsten Fakten zum Ewing-Sarkom sind in der folgenden Übersicht kurz zusammengefasst. Ausführliche Informationen zu dieser seltenen, schweren Erkrankung finden sich im nachfolgenden Artikel.
- Definition: Die Ewing-Sarkome stellen die zweithäufigste Form der seltenen primären bösartigen Knochentumore im Kindes- und Jugendalter dar. Unter dieser Bezeichnung werden verschiedene aggressive, hochmaligne und metastasierende Tumore zusammengefasst, die aus primitiven Zellen entstehen.
- Symptome: Die typischen Beschwerden sind eher unspezifisch und äußern sich durch Schmerzen und Schwellungen im betroffenen Knochenbereich. Am häufigsten liegt der Krankheitsursprung in den langen Röhrenknochen, aber es können alle Knochen befallen werden. Zudem neigt der Tumor zur Metastasenbildung. Ist dies der Fall treten häufig die typischen Anzeichen einer Krebserkrankung auf.
- Ursachen: Die Entstehung dieser Sarkome ist noch ungeklärt. Es wird vermutet, dass primitive Zellen mit einer bestimmten Chromosomentranslokation das Tumorwachstum auslösen. In den meisten Fällen bildet sich der Tumor im (wachsenden) Knochengewebe, selten in den Weichteilen.
- Diagnose: Bei der Diagnose kommen bildgebende Verfahren zum Einsatz, die ein Ewing-Sakrom in vielen Fällen aufgrund spezifischer Strukturen erkennen lassen. In jedem Fall muss die Diagnose durch eine Biopsie gesichert werden. Weitere Untersuchungen dienen der Suche nach möglichen Metastasen und der Differenzialdiagnostik.
- Behandlung: Die heutige Standardtherapie setzt sich aus einer Induktionschemotherapie, einer Lokaltherapie mittels Operation und/oder Bestrahlung und einer abschließenden adjuvanten Chemotherapie zusammen. Die Behandlung eines wiederkehrenden Tumors ist prinzipiell schwieriger.
- Prognose: Besteht nur ein lokaler Tumor ohne Metastasen ist derzeit laut Statistik eine langfristige Heilung bei etwa 65 Prozent der Betroffenen zu erwarten. Haben sich bereits Metastasen gebildet oder tritt der Krebs nach erfolgreicher Behandlung erneut auf, verschlechtert dies die Prognose deutlich.
- Aktueller Forschungsstand: Neuste Erkenntnisse konnten die Prognosen und Überlebenschancen deutlich verbessern. Die schwere Erkrankung ist weiterhin Bestandteil vieler Forschungsansätze um Betroffenen immer bessere Behandlungsoptionen anbieten zu können.

Definition
Sarkome sind seltene bösartige Krebserkrankungen, die ihren Ursprung im Binde- und Stützgewebe (Knochen, Knorpel und Fettgewebe) oder im Muskelgewebe haben können. Das sogenannte Ewing-Sarkom (auch Ewing Sarkom) bezeichnet einen primären bösartigen Knochentumor, der hauptsächlich bei Kindern und Jugendlichen auftritt. Neben dem Osteosarkom, stellen die Ewing-Sarkome die zweithäufigsten Knochentumore im jungen Lebensalter. Bei circa zehn bis fünfzehn Prozent aller malignen Knochentumore wird ein solches Sarkom diagnostiziert. Die Krankheit manifestiert sich insbesondere im Alter zwischen zehn und zwanzig Jahren, wobei der Gipfel bei zwölf bis siebzehn Jahren liegt. Seltener tritt die Erkrankung bei jüngeren Kindern, Säuglingen oder im (jungen) Erwachsenenalter auf. Tendenziell erkranken etwas mehr Jungen als Mädchen.
Nach den aktuellen Kriterien der WHO (Weltgesundheitsorganisation) werden unter den Ewing-Sarkomen verschiedene Tumore zusammengefasst, die ursprünglich gesondert klassifiziert wurden. Der Begriff umfasst demnach alle bisher beschriebenen Sarkome aus der Gruppe der Ewing-Tumore, die Askin-Tumore der Thoraxwand, die atypischen Ewing-Sarkome sowie alle Formen maligner peripherer neuroektodermaler Tumore (PNET oder MPNET). Ewing-Sarkome sind aggressive (hochmaligne) und metastasierende Tumore, die aus primitiven, kleinen, runden Zellen entstehen. Diese lassen sich in der Diagnostik blau einfärben („kleine blaue Zellen“).
Namensgebend für die Erkrankung war der US-amerikanische Pathologe James Ewing (1866-1943), der sich unter anderem in der Krebsforschung engagierte. Bestehende Vorarbeiten und Beschreibungen ähnlicher Tumore von Georg Albert Lücke (1829-1894) und Otto Hildebrand (1858-1927), ließen ihn im Jahr 1920 mit der Beschreibung des damals noch neuartigen Knochentumors einen medizinischen Durchbruch erlangen.
Wichtig ist die Abgrenzung eines Knochensarkoms von einer Knocheninfektion beziehungsweise von einer Knochenentzündung (Osteitis) oder Knochenmarkentzündung (Osteomyelitis).
Symptome
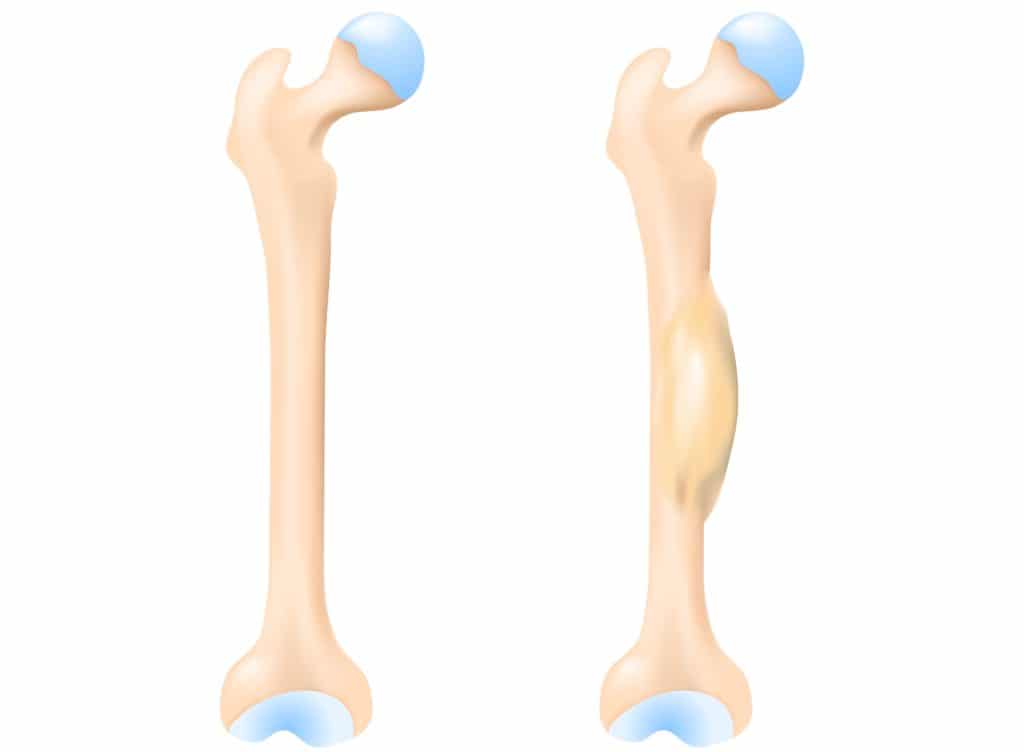
Die typischen Beschwerden sind unspezifisch und äußern sich vor allem durch Schmerzen und möglicherweise auch über eine Schwellung im Knochenbereich, die ertastet werden kann. Generell können alle Knochen betroffen sein, am häufigsten ist der Krebs aber im Knochenschaft (Diaphyse) der langen Röhrenknochen von Oberschenkel und Oberarm, sowie im Becken, in den Rippen und im Schlüsselbein zu finden.
Die Lokalisation und Art des Schmerzes ist davon abhängig welche Bereiche betroffen sind. Befindet sich der Tumor beispielsweise im Becken, kann es zu tiefen Kreuzschmerzen oder Rückenschmerzen kommen. Liegt der Ursprungsort in den Rippen, kann es zu Flankenschmerzen kommen. Dabei können die Schmerzen auch intermittierend auftreten und unter Belastung zunehmen.
Aufgrund der Tumorausbreitung werden befallene Knochen instabil und Knochenbrüche können die Folge sein. Des Weiteren können auch typische Anzeichen für Krebs im Allgemeinen auftreten, wie etwa eine Lymphknotenschwellung, Nachtschweiß, Fieber, herabgesetzte Leistungsfähigkeit und Gewichtsverlust. Treten diese Beschwerden auf, liegt häufig bereits eine Absiedelung des Krankheitsherds in andere Körperteile vor. Bei etwa einem Viertel der Erkrankten wird bereits bei der Erstdiagnose einer Metastasierung (Bildung von Tochtergeschwülsten) nachgewiesen. Oft finden sich Metastasen in der Lunge oder in anderen Knochen und im Knochenmark.
Ursachen
Die genaue Entstehung des Ewing-Sarkoms ist bis heute noch unklar. Es wird davon ausgegangen, dass die Erkrankung von primitiven Zellen ausgeht, die eine bestimmte genetische Abnormalität aufweisen. Dabei handelt es sich zumeist um eine Chromosomenmutation beziehungsweise um eine Translokation zwischen den Chromosomen 11 und 22. Das dabei neu entstehende Gen wird EWS/FLI1 genannt und führt in seiner Funktion wahrscheinlich zur Krebsbildung.
Eine genetische Prädisposition ist nicht anzunehmen, aber es fällt auf, dass so gut wie keine Krankheitsfälle in der afrikanischen und chinesischen Bevölkerung bekannt sind. Da sich der Tumor hauptsächlich bei Heranwachsenden bildet, liegt die Vermutung nahe, dass bevorzugt wachsende Knochensubstanz befallen wird.
In den allermeisten Fällen ist diese Krebsart knöchernen Ursprungs, seltener (und eher im höheren Erkrankungsalter) kann ein Ewing-Tumor aber auch extraossär – als sogenanntes Weichteilsarkom – auftreten.

Diagnose
Durch die unspezifische Symptomatik und möglichen Verwechselungen mit anderen Erkrankungen wie etwa einer Knochenentzündung kommt es nicht selten zu einer Verzögerung bei der Diagnose.
Besteht die Vermutung auf eine Erkrankung der Knochen, werden in aller Regel zunächst Röntgenaufnahmen gemacht. Auf den Röntgenbildern lässt sich ein knöchernes Ewing-Sarkom durch mottenfraßähnliche Knochenschäden erkennen, gegebenenfalls auch mit Infiltration umgebener Gewebe. Auch die Knochenhaut (Periost) kann betroffen sein, was sich in verschiedenen Periostreaktionen zeigt. Beispiele sind die zwiebelschalenartige Abhebung mehrerer Lamellen, der Periostsporn (Codman-Dreieck) und sogenannte Spiculae als sonnenstrahlähnliche Strukturen.
Mit Hilfe anderer bildgebender Verfahren (Computertomographie und Magnetresonanztomographie), können oftmals präzisere Aussagen zum Tumor und zum Weichteilgewebe getroffen werden.
Besteht aufgrund dieser Untersuchungen der Verdacht auf ein Ewing-Sarkom, wird zur Diagnoseabsicherung immer eine Gewebeprobe des Tumors für weitere Untersuchungen entnommen (Biopsie). Dabei ist es im Hinblick auf spätere Therapiemaßnahmen besonders wichtig, dass mögliche operative Zugangswege zum Tumor berücksichtigt beziehungsweise genutzt werden. Eine Biopsie sollte demnach nur von Ärzten durchgeführt werden, die auf Sarkome spezialisiert sind. Der spätere Operateur sollte in jedem Fall frühzeitig in die Diagnosestellung mit einbezogen werden, um das Risiko von Komplikationen zu minimieren.
Bei der Tumorgewebeuntersuchung ermöglichen immunhistochemische (Antikörperfärbung) und morphologische Befunde, möglicherweise ergänzt durch den genetischen Nachweis einer Chromosomentranslokation, eine sichere Aussage über das Vorliegen der Erkrankung. Die Ergebnisse sichern außerdem eine differenzialdiagnostische Abgrenzung, etwa zu anderen klein-, blau-, rundzelligen Tumoren oder einer Osteomyelitis. Immunhistochemisch ergibt sich bei einem Ewing-Sarkom eine positive Färbung (blau) für den Antikörper O13 (CD99).
Da sich diese Art von Tumor häufig über Metastasen ausbreitet, schließt sich direkt nach der Erstdiagnose auch eine Ausbreitungsdiagnostik (Staging) an, bei der zunächst die häufigsten Lokalisationen von Metastasen überprüft werden. Dabei kommen erneut bildgebende Verfahren zum Einsatz, zum Beispiel zur Untersuchung der Lunge. Auch werden eine Knochenszintigraphie sowie eine Knochenmarkbiopsie und ergänzende Knochenmarkaspiration zum Nachweis von Tumorzellen durchgeführt. Unter Umständen kommt eine Lumbalpunktion in Frage. Der Vorteil einer zusätzlichen nuklearmedizinischen Ganzkörper-Positronen-Emissions-Tomographie (PET) wird noch diskutiert.
Behandlung
Die Ewing-Sarkome sind äußert aggressive, hochmaligne Tumore, an denen die allermeisten Betroffenen ohne eine entsprechende Therapie versterben würden. Medizinische Fortschritte in der Behandlung haben es möglich gemacht, dass die Krankheit besser behandelbar und unter gewissen Voraussetzungen sogar heilbar ist. Dabei folgt die Behandlung von Knochen- und Weichteil-Sarkomen dem gleichen Vorgehen.
Am wirksamsten hat sich bisher die Kombination aus einer medikamentöse Behandlung (Chemotherapie) und einer Lokaltherapie (Operation und Bestrahlung) erwiesen. Das Standardverfahren sieht dabei zunächst eine neoadjuvante Chemotherapie (Induktionschemotherapie) vor, die einer Metastasierung vorbeugen und durch eine Verringerung der Tumormasse die weitere Behandlung ermöglichen soll. Anschließend kommt es bei der Lokaltherapie zu einer operativen Entfernung und/oder radiotherapeutischen Behandlung des Tumors und möglicher Metastasen. Mittels einer adjuvanten Chemotherapie sollen zum Abschluss der Therapie Krebszellen abgetötet werden, die möglicherweise noch im Körper verblieben sind.
Bisher werden, oft auch in Kombination, die Substanzen Actinomycin D, Cyclophosphamid, Doxorubicin, Etoposid, Ifosfamid und Vincristin zur medikamentösen Therapie angewandt. Häufig kommt es zu schweren Nebenwirkungen, vor allem bei älteren Betroffenen. Der Einsatz von Hochdosischemotherapien mit Stammzelltransplantation, Bisphosphonaten und andere Methoden werden bisher noch getestet.
Bei der Operation entstehende Knochendefekte können mittels unterschiedlicher Rekonstruktionsverfahren im Nachgang versorgt werden, wie zum Beispiel mit (Endo-) Prothesen. Oft schließt sich der Behandlung, die bis zu ein Jahr andauern kann, eine Rehabilitation an.
Ewing-Sarkome zeigen eine Tendenz zum Wiederauftreten, was einen wichtigen Aspekt bei der Behandlung darstellt. Anhand verschiedener Kriterien (Ewing 2008-Studie) werden die Betroffenen heutzutage gemäß des Standardvorgehens in drei Rezidiv-Risikogruppen eingeteilt, aus denen die jeweilige Intensität der Chemotherapie (Anzahl der Zyklen) abgeleitet wird
Die relativ hohe Rezidivrate erfordert regelmäßige Nachsorgeuntersuchungen, bei denen nicht nur die Entstehung am gleichen Ursprungsort sondern auch mögliche Metastasenbildungen kontrolliert werden. Die Behandlung bei einem erneuten Auftreten ist zumeist die Intensivierung der Chemotherapie. Weitere Ansätze befinden sich noch in der Testphase.
Naturheilkundliche Behandlung
Sarkome bedürfen aufgrund ihrer Aggressivität und des schnellen Metastasierens einer speziellen schulmedizinischen Behandlung. Wie bei jeder anderen Krebserkrankung kann aber eine biologische Krebstherapie dabei helfen, Körper und Psyche zu stärken. In Absprache mit den behandelnden Ärzten und unter Berücksichtigung des individuellen Krankheitsverlaufs können gegebenenfalls verschiedene Naturheilverfahren den Heilungsprozess unterstützen.
Prognose
Handelt es sich ausschließlich um einen lokalen Tumor ohne Metastasen besteht eine eher positive Prognose bei der Ersterkrankung. Ist dies der Fall können derzeit etwa 65 Prozent der Betroffenen langfristig geheilt werden. Bei circa fünfzehn Prozent der Betroffenen treten allerdings bereits bei der Erstdiagnose Metastasen auf, was die Heilungschancen verschlechtert. Laut Statistik wird dann von einer Fünf-Jahres-Überlebensrate bei durchschnittlich einem Viertel der Erkrankten ausgegangen. Ist nur die Lunge betroffen, bestehen zumeist bessere Chancen als bei einer weiteren Ausbreitung in andere Körperregionen.
Tritt der Krebs erneut auf, verschlechtert sich die Prognose ähnlich ungünstig. Der wichtigste Faktor ist in diesem Zusammenhang der Zeitraum zwischen der Erstdiagnose und dem Wiederauftreten. Je nach Patientenfall können die Heilungschancen aber von den statistischen Werten abweichen.
Aktueller Forschungsstand
Die neusten medizinischen Erkenntnisse wurden in der Leitlinie „Ewing-Sarkome des Kindes- und Jugendalters“ zusammengefasst und stellen unter anderem den aktuellen Behandlungsstandard. Die neuen Therapieansätze konnten zeigen, dass sich die langfristigen Heilungschancen für Patienten mit einem lokal begrenzten Ewing-Sarkom auf 60 bis 70 Prozent verbessert haben. Einer im Journal of Clinical Oncology veröffentlichten Studie zufolge, können Überlebende mit nur wenigen Einschränkungen zu einem weitestgehend normalen Leben zurückkehren.
Für weitere Verbesserungen in der Therapie und neue Behandlungsoptionen finden viele klinische Studien statt. Getestet werden zum Beispiel die neue Medikamente Irinotecan und Temozolomid, die in Kombination und besonders bei wiederkehrenden Ewing-Sarkomen Wirksamkeit zeigen. Dies haben kürzlich Forscher im Fachjournal Acta Oncologica bekanntgegeben.
Weitere Arbeiten konzentrieren sich auf die Erforschung und Zuordnung von Tumoren, die dem Ewing-Sarkom sehr ähnlich sind (Ewing-like Sarkome). (tf, cs)
Zum Weiterlesen:
Knochenschmerzen
Knochenmarkentzündung
Autoren- und Quelleninformationen
Dieser Text entspricht den Vorgaben der ärztlichen Fachliteratur, medizinischen Leitlinien sowie aktuellen Studien und wurde von Medizinern und Medizinerinnen geprüft.
- Grevener, Knut / Haveman, Lianne M. / Ranft, Andreas / u.a.: Management and Outcome of Ewing Sarcoma of the Head and Neck; Pediatric blood & cancer, Volume 63, Issue 4, April 2016, Pediatric blood & cancer
- Gesellschaft für Pädiatrische Onkologie und Hämatologie (GPOH): S1-Leitlinie Ewing-Sarkome des Kinder- und Jugendalters, Stand: Juni 2014, Leitlinien-Detailansicht
- Amboss GmbH: Bösartige Knochentumoren (Abruf: 26.06.2019), amboss.com
- Mayo Clinic: Ewing sarcoma (Abruf: 26.06.2019), mayoclinic.org
- National Cancer Institute (NCI): Ewing Sarcoma Treatment (Abruf: 26.06.2019), cancer.gov
- American Academy of Orthopaedic Surgeons: Ewing's Sarcoma (Abruf: 26.06.2019), orthoinfo.aaos.org
- UpToDate, Inc.: Clinical presentation, staging, and prognostic factors of the Ewing sarcoma family of tumors (Abruf: 26.06.2019), uptodate.com
- National Health Service UK: Ewing sarcoma (Abruf: 26.06.2019), nhs.uk
- U.S. National Library of Medicine (NLM): Ewing sarcoma (Abruf: 26.06.2019), ghr.nlm.nih.gov
- Ranft, Andreas / Seidel, Corinna / Hoffmann, Christiane / u.a.: Quality of Survivorship in a Rare Disease: Clinicofunctional Outcome and Physical Activity in an Observational Cohort Study of 618 Long-Term Survivors of Ewing Sarcoma, Journal of Clinical Oncology, 2016, ascopubs.org
Wichtiger Hinweis:
Dieser Artikel enthält nur allgemeine Hinweise und darf nicht zur Selbstdiagnose oder -behandlung verwendet werden. Er kann einen Arztbesuch nicht ersetzen.





