

Glasknochen – Osteogenesis imperfecta
Die Osteogenesis imperfecta bedeutet „unvollkommene Knochenbildung“ und wird umgangssprachlich auch als Glasknochenkrankheit bezeichnet. Diese seltene Erbkrankheit beruht auf bestimmten Gendefekten, die den Kollagenhaushalt beeinflussen. Damit kommt es bei den Betroffenen zu Störungen im Bindegewebe und im Knochenstoffwechsel. Die Folge verformbarer und leicht zerbrechlicher Kochen stellt das Leitsymptom dar, welches hinter dem bildhaften Begriff der Glasknochen steht. Die Behandlung stützt sich ausschließlich auf symptomatische Therapiemethoden, um Knochenfrakturen möglichst zu verhindern und bestmöglich zu versorgen.
Inhaltsverzeichnis
Ein kurzer Überblick
In der folgenden Übersicht sind die wichtigsten Fakten rund um das Thema der seltenen Erbkrankheit Osteogenesis imperfecta aufgeführt. Ausführliche Informationen zu dem komplexen Krankheitsbild finden sich in dem nachfolgenden Artikel.
- Definition: Die Osteogenesis imperfecta (Glasknochen, Glasknochenkrankheit) bezeichnet eine erblich bedingte Kollagenfehlbildung, die in erster Linie eine Bindegewebsstörung hervorruft. Namentlich kommt es bei der Erkrankung zu einer unvollkommenen Knochenbildung, was sich über die Leitsymptome vermehrter Knochenbrüche und Knochenverformungen äußert.
- Symptome: Neben der abnorm hohen Anfälligkeit für Spontanfrakturen und Knochenverformungen verursacht die Krankheit eine Vielzahl weiterer komplexer Beschwerdebilder. Zu den typischen Symptomen zählen unter anderem Muskelschwäche und blaue Skleren. Der Schwergrad der Erkrankung reicht von sehr leichten Formen bis hin zu letalen Ausprägungen.
- Ursachen: Auslöser sind verschiedene Genmutationen, die für Störungen in der Kollagenbildung (Kollagen Typ 1) verantwortlich sind. Kollagen ist hauptsächlicher Bestandteil des Bindegewebes und ein wichtiger Baustein für die Knochen und andere Strukturen und Gewebe. Daher kommt es nicht nur zu Störungen im gesamten Bewegungsapparat sondern auch in vielen anderen Körperbereichen.
- Diagnose:Neben einer gründlichen Anamnese und klinischen Untersuchung kommen vor allem Röntgenuntersuchungen bei der Diagnosestellung zum Einsatz. Eine Absicherung der Diagnose kann gegebenenfalls über einen genetischen Nachweis erfolgen. Vorgeburtlich sind schwerere Krankheitsformen oft mittels pränatalem Ultraschall erkennbar.
- Behandlung: Die schulmedizinische Behandlung der Symptome stützt sich zumeist auf relativ neue Medikamente (Biophosphonate), orthopädische Maßnahmen und eine kontinuierliche Physiotherapie. Die Therapiemöglichkeiten befinden sich noch in der Entwicklung. Ergänzend können allgemeine Maßnahmen zur Gesundheitsförderung und eine osteopathische Behandlung hinzugezogen werden.
- Aktueller Forschungsstand: Aktuelle Forschungen zu den Krankheitsursachen (Molekulargenetik) und Therapiemöglichkeiten sollen die Kenntnisse zu dieser seltenen Erbkrankheit erweitern und Betroffenen eine bestmögliche Versorgung sicherstellen.
Definition
Der medizinische Fachbegriff Osteogenesis imperfecta (allgemeine Abkürzung: OI) leitet sich aus dem Griechischen ab und bezeichnet eine unvollkommene Knochenbildung. Im allgemeinen Sprachgebrauch werden aber zumeist die Begriffe Glasknochen oder Glasknochenkrankheit verwendet, die sich auf das Hauptsymptom vermehrter Knochenbrüche beziehen. In erster Linie handelt es sich bei dem Krankheitsbild aber um eine seltene erblich bedingte Bindegewebsstörung, die aufgrund verschiedener Gendefekte auftritt und zu einer Kollagenfehlbildung (Kollagen Typ I) führt. Die Osteogenesis imperfecta tritt bei circa einer von 20.000 Geburten auf.
Klassifizierung und klinische Merkmale
Eine Vielzahl an verschiedenen Gendefekten und dadurch verursachte Störungen in der Kollagenzusammensetzung führen zu dieser Erkrankung. Aufgrund dessen finden sich auch unterschiedliche Beschwerdebilder bei den Betroffenen und das Krankheitsbild zeigt eine große Variabilität im Schweregrad und im Krankheitsverlauf.
Im Jahr 1979 wurde erstmals eine Vier-Typen-Klassifizierung eingeführt, die verschiedene Krankheitsformen und ihre klinischen Besonderheiten unterteilt. Die Beschreibung der Typen I bis IV geht auf den australischen Mediziner David Sillence zurück. Bei diesen vier Formen treten Mutationen der Kollagen Typ 1 kodierenden Gene auf (COL1A1 oder COL1A2), welche in Europa zu mehr als 80 Prozent ursächlich sind für die Erkrankung.
Fortlaufend neue genetische und klinische Erkenntnisse haben zu einer Erweiterung dieser Aufteilung geführt. Aktuell sind elf Typen beschrieben, bei denen auch noch weitere Gene und Mutationsformen berücksichtigt sind. Allerdings finden nur die Typen I bis VI eine weitestgehend einheitliche Verwendung. Bis heute können noch nicht alle Ausprägungen zugeordnet werden, denn die Vielzahl an ursächlichen Genen erschwert eine eindeutige Klassifizierung. Auch die erweiterte Klassifizierung ist daher noch immer in Diskussion und es besteht weiterhin Bedarf bei der Aufklärung unklassifizierter Krankheitsformen.
Der häufige Typ I mit einer leichten Verlaufsform ist auch unter der Bezeichnung Osteogenesis imperfecta tarda oder Typ Lobstein bekannt und die schwerwiegendste, letale Form vom Typ II wird als Osteogenesis imperfecta congenita beziehungsweise Typ Vrolik bezeichnet.

Symptome
Trotz der hohen Variabilität bei den auftretenden Symptomen, steht in nahezu allen Krankheitsfällen die abnorm hohe angeborene Anfälligkeit der Knochen für Brüche (Osteopsathyrose) im Vordergrund. Die Knochenbrüche treten infolge einer reduzierten Knochenmasse und mangelnder Stabilität und Elastizität ohne adäquate Traumata auf (Spontanfrakturen).
Darüber hinaus kommt es auch zu (ausgeprägten) Skelettdeformierungen, insbesondere der langen Röhrenknochen in den Extremitäten und der Wirbelsäule (Skoliose, Kyphose) und zu Kleinwuchs.
Neben diesen Hauptsymptomen kann es je nach Ausprägung unter anderem zu folgenden weiteren Symptomen kommen:
- Muskelschwäche, verringerte Muskelspannung (Muskelhypotonie),
- überdehnbare Gelenke und Bänder,
- eigenständige Knocheninseln (Schaltknochen) an der Schädeldecke,
- blaue Skleren (Lederhäute der Augen),
- Schwerhörigkeit,
- Kurzsichtigkeit,
- Herzprobleme,
- Lungenstörungen (Verformung des Brustkorbs),
- Verfärbungen und Brüchigkeit der Zähne (Dentinogenesis imperfecta),
- Neigung zu Hämatomen,
- weiche, durchscheinende Haut,
- dreieckige Gesichtsform mit breiter Stirn und abstehenden Ohren.
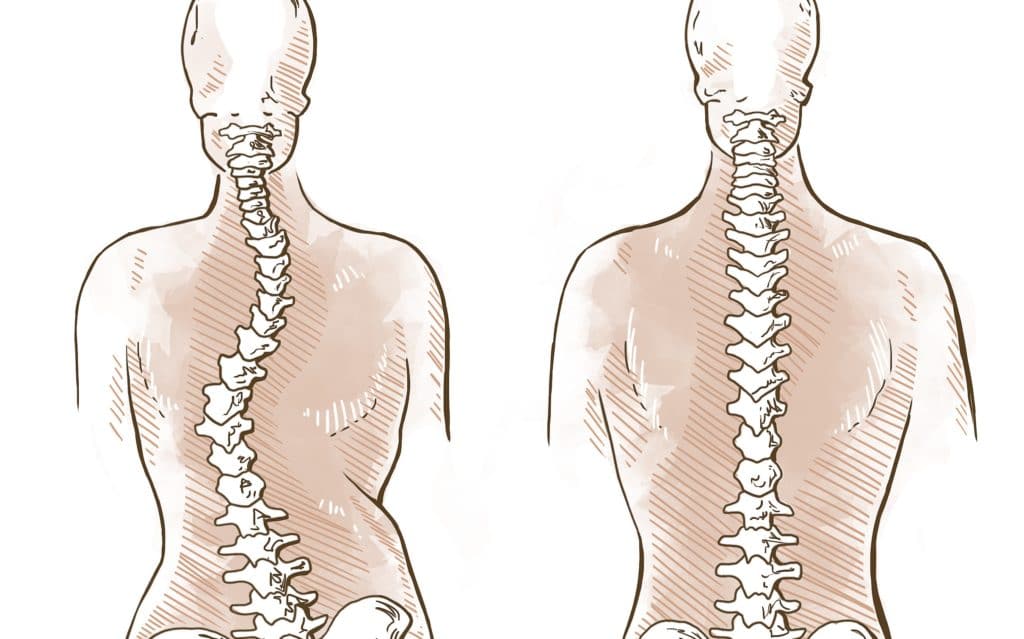
Krankheitsverlauf
Die vielen möglichen Beschwerden und Schweregrade bedingen individuell sehr unterschiedliche Krankheitsverläufe. Bei mittleren und schweren Krankheitsverläufen treten bereits während der ersten Lebensmonate Frakturen und Deformierungen an den Extremitäten und der Wirbelsäule auf. Besonders kritisch sind die Wachstumsphasen bis ins Jugendalter, während derer bereits unter sehr geringer Krafteinwirkung immer wieder Frakturen auftreten.
Aber auch vorgeburtlich können schon Frakturen, Deformierungen und andere Auffälligkeiten an den Knochen auftreten. Bei der schwerwiegendsten Form (Typ II) verläuft die Krankheit entweder vorgeburtlich letal oder es ist in den ersten Lebensmonaten mit einem tödlichen Verlauf zu rechnen. Weitere schwerwiegende Formen können, aufgrund der Häufigkeit auftretender Frakturen und möglicher Knochenverbiegungen, die Betroffenen am Stehen und Gehen hindern.
Besteht hingegen eine leichte Form der Erkrankung (Typ I) erfahren Betroffene meist nur wenige Frakturen in ihrer Kindheit und Jugend ohne weitere Einschränkungen. Viele Krankheitsformen haben gemeinsam, dass die Frakturrate nach dem Ende der Pubertät deutlich zurückgeht.
Prinzipiell besteht bei den lebensfähigen Formen und unter entsprechenden Therapieanwendungen keine verminderte Lebenserwartung.
Ursachen
Die seltene Erbkrankheit wird vorranging autosomal-dominant vererbt. Eine Vererbung findet demnach unabhängig vom Geschlecht statt und die Krankheit kann sich bereits dann manifestieren, wenn sich der entsprechende Gendefekt nur auf einem der 22 Autosomenpaare (Körperchromosomen) befindet. Wenn keiner der beiden Elternteile die Krankheit übertragen hat, kommt darüber hinaus eine Spontanmutation als Auslöser in Frage.
Die häufigsten genetischen Mutationen, die dieser Erkrankung zugeordnet werden, führen zu quantitativen oder qualitativen Störungen in der Kollagenbildung. Meistens ist das Kollagen Typ I betroffen, welches dem allgemeinen Kollagen-Begriff oft gleichgesetzt wird. Kollagene sind die häufigsten faserbildenden Eiweiße (Strukturproteine, Faserproteine) mit bindenden und stützenden Funktionen in verschiedenen Teilen des menschlichen Körpers. Kollagen Typ I ist hauptsächlicher Bestandteil des Bindegewebes. Ebenso ist es ein wichtiger Baustein für Knochen, Knorpel, Sehnen, Bänder, Zähne, und Haut sowie die Bindehaut (Konjunktiva). Die Erkrankung wirkt sich also nicht nur auf das Skelettsystem aus, sondern auf den gesamten Bewegungsapparat und auf alle weiteren Strukturen, die zu einem gewissen Anteil Kollagen Typ 1 enthalten.
Trotz der eindeutigen Ursache der Genmutation, ist diese selbst mit den neuesten Methoden nicht in jedem Fall genetisch nachweisbar.
Diagnose
Die ersten Anhaltspunkte bei der Diagnosestellung liefert die Patientenbefragung (Anamnese), bei der neben den typischen Symptomen auch ein familiäres Vorkommen einer Osteogenesis imperfecta abgeklärt wird. Ergänzend kommen körperliche Untersuchungen, Röntgenaufnahmen und Laboruntersuchungen zum Einsatz, um auch andere mögliche Skeletterkrankungen auszuschließen, die im Kindesalter auftreten (zum Beispiel Rachitis und Hypochondroplasie). Besondere Bedeutung kommt dem Ausschluss von ursächlichen Traumata für vorliegende Knochenfrakturen zu. Dies kann auch bei Verdachtsfällen auf Kindesmisshandlung relevant werden.
Im Röntgenbild lässt sich eine erhöhte Transparenz der Knochen erkennen. Die Strukturen erscheinen glasartig, da zu wenig schattengebende Knochensubstanz vorhanden ist. Die äußere Schicht ist meist strichförmig verdünnt. Häufig wird auch eine Kallusbildung sichtbar. Dabei handelt es sich um Narbengewebe, welches den Knochen an der Bruchstelle verbreitert und deformiert.
Zudem kann eine Knochendichtemessung Aufschluss über die Erkrankung geben, denn bei Betroffenen ist die Knochendichte deutlich vermindert. Eine zuverlässige Beschreibung der Knochenstruktur und der Muskel-Knochen-Interaktion ist über eine Computertomographie möglich, die aber nicht bei jedem Betroffenen anwendbar ist (Mindestkörpergröße).
Treten neben den Symptomen am Skelett auch Beschwerden an anderen Strukturen und Organen auf, werden auch diese klinisch untersucht.
Eine Absicherung und Erweiterung der Diagnose bietet eine genetische Untersuchung, bei der nach ursächlichen Genmutationen gesucht wird. Zwar ist es nicht in jedem Fall möglich die Erkrankung und den zugrundeliegenden Gendefekt nachzuweisen, aber eine mögliche Identifizierung dient neben der Diagnosefindung auch der weiteren Klassifizierung und Aufklärung des eigenen Weitervererbungsrisikos. Wurde ein Gennachweis erbracht, bedeutet dies aber nicht gleichzeitig, dass auch eine zuverlässige Aussage zur individuellen Ausprägung oder Lebensfähigkeit getroffen werden kann. Vor der Geburt wird eine zytogenetische Untersuchung mit Hilfe einer Mutterkuchenpunktion (Chorionzottenbiopsie) durchgeführt.
Generell können die schwerwiegenderen Formen der Glasknochenkrankheit im Mutterleib bereits während eines pränatalen Ultraschalls nachgewiesen werden. Typische Befunde sind unter anderem verkürzte und deformierte Extremitätenknochen, Rippenfrakturen und gelegentlich auch Kallusbildungen.

Behandlung
Bislang gibt es keinerlei Möglichkeiten für eine Ursachenbekämpfung oder Heilung dieser Erbkrankheit. Die symptomatische Behandlung stütz sich auf die drei schulmedizinisch anerkannten Pfeiler der medikamentösen Behandlung, der orthopädischen Behandlung und der physiotherapeutischen Behandlung.
Medikamentöse Behandlung
In den letzten Jahren haben sich vor allem bei mittleren und schweren Fällen intravenös verabreichte Bisphosphonate als erfolgreiche Therapiemöglichkeit erwiesen. Die Medikamente (zum Beispiel Neridronat und Pamidronat) bewirken eine Zunahme der Knochenmasse und eine Steigerung der Knochenfestigkeit. Dadurch kommt es seltener zu Knochenbrüchen und einer verbesserten Beweglichkeit. Noch haben die verschiedenen Bisphosphonate keine Zulassung für die Therapie von Osteogenesis imperfecta, weshalb eine Anwendung individuell geklärt werden muss. Nur unter ausdrücklicher Einwilligung der Betroffenen kann ein sogenannter „individueller Heilversuch“ mit diesen Arzneimitteln erfolgen. Engmaschige Kontrolluntersuchungen zur Wirksamkeit und zu potenziellen Nebenwirkungen sind unabdingbar.
Entgegen einiger Behandlungskonzepte anderer Knochenerkrankungen ist eine Zufuhr von Vitamin D und Kalzium über die allgemeinen Empfehlungen hinaus nicht sinnvoll.
Orthopädische Behandlung
Die am häufigsten auftretenden Symptome von Knochenbrüchen und Knochenverformungen erfordern in der Regel intensive orthopädische Behandlung. Das Ziel dieser Therapieform ist vor allem die Funktionalität und die Belastbarkeit des Skelettsystems zu erhalten oder wiederherzustellen. Konservative Maßnahmen bei gebrochenen Extremitätenknochen umfassen vor allem bei sehr kleinen Kindern spezielle Techniken der Wicklung und der Lagerung. Des Weiteren kommt es zum Einsatz von Orthesen (zum Beispiel Schienen) und Gipsverbänden.
In komplizierteren Fällen werden aber auch operative Eingriffe notwendig, um Frakturen und Fehlstellungen zu vermeiden oder aber zu korrigieren und zu heilen. Bei Operationen im Kindes- und Jugendalter ist eine Berücksichtigung der Wachstumsphasen von besonderer Bedeutung. Dies erfordert nach Möglichkeit die Verwendung eines Teleskopnagelsystems (Bailey- oder Fassier-Duval-Nagel). Dabei werden, oft nach mehreren gezielten Knochendurchtrennungen (Osteotomien), zwei ineinandergeschobene Nagelteile in das Innere des Knochens eingebracht. Die Knochenteile werden dadurch wieder verbunden und während des Wachstums gleiten diese Nägel zusammen mit dem Knochen auseinander. So können die Knochen über einen langen Zeitraum stabilisiert werden.
Bei sehr kleinen Kindern kann es sein, dass die Knochen noch nicht ausreichend Raum für einen Teleskopnagel bieten. Ist dies der Fall, kommen andere Hilfsmittel zum Einsatz, die dann gegebenenfalls zu einem späteren Zeitpunkt entfernt oder ausgewechselt werden müssen.
Physiotherapeutische Behandlung
Um der Gefahr von Fehlstellungen, Fehlhaltungen und Skelettveränderungen vorzubeugen ist eine individuell abgestimmte Physiotherapie von großer Bedeutung. Aber auch bei der Rehabilitation nach Knochenfrakturen ist eine gezielte Krankengymnastik zentraler Bestandteil der Therapiemaßnahmen. Das vorrangige Ziel ist es, die Beweglichkeit zu verbessern oder zu erhöhen und die Muskulatur zu stärken. Jeder Betroffene sollte eine regelmäßige und kontinuierliche physiotherapeutische Behandlung erhalten und die anderen Behandlungsmaßnahmen dadurch unterstützen.
Sind weitere Körperbereiche oder Organe betroffen, wie beispielsweise die Lunge oder das Herz, können zusätzliche Therapieformen notwendig werden.
Naturheilkundliche Behandlung
Die schulmedizinischen Verfahren zur Behandlung einer Osteogenesis imperfecta können durch allgemeine, gesundheitsfördernde Maßnahmen unterstützt werden. So wirken sich möglichst viel Bewegung, Sport (Schwimmen) und ein Verzicht auf Zigaretten und Alkohol positiv aus.
Neben der physiotherapeutischen Behandlung können auch Maßnahmen aus dem Bereich der Osteopathie zur Linderung der Beschwerden beitragen. Ein Besuch beim Heilpraktiker kann ebenso hilfreich sein.
Aktueller Forschungsstand
Obwohl die Krankheit bereits seit langem bekannt ist, erfordern die komplexen genetischen Grundlagen und stark variierenden Krankheitsausprägungen intensive Forschung, besonders im Bereich der Krankheitsursachen und der individuellen Therapiemöglichkeiten.
Im 2017 veröffentlichten Konsensuspapier der „Monatszeitschrift Kinderheilkunde“ werden Studien und Forschungsansätze im Bereich der Anwendung von Biophosphonaten und anderen Medikamenten erwähnt wie beispielsweise die Auswirkungen von Parathormon als knochenaufbaustimulierendes Medikament bei Erwachsenen. Auch für den Bereich der Gen- und Zelltherapie als Behandlungsoption werden internationale Studien durchgeführt.
Erst vor kurzem wurde die Veränderung des Kollagens entschlüsselt, die sehr wahrscheinlich bei der Glasknochenkrankheit für die verringerte Widerstandsfähigkeit der Knochen verantwortlich ist.
Da es sich um ein seltenes Krankheitsbild handelt gibt es noch keine definierten Leitlinien für die Betreuung der Betroffenen. In der Fortbildungszeitschrift „pädiatrie hautnah“ haben deutsche Fachärzte, die seit langem Kinder und Jugendliche mit Osteogenesis imperfecta betreuen, den aktuellen Stand zum Krankheitsbild zusammengefasst. (tf, cs)
Autoren- und Quelleninformationen
Dieser Text entspricht den Vorgaben der ärztlichen Fachliteratur, medizinischen Leitlinien sowie aktuellen Studien und wurde von Medizinern und Medizinerinnen geprüft.
- Deutsche Gesellschaft für Osteogenesis imperfecta (Glasknochen) Betroffene e.V.: Informationen für Betroffene (Abruf: 03.07.2019), oi-gesellschaft.de
- Merck & Co., Inc.: Osteogenesis imperfecta (Abruf: 03.07.2019), msdmanuals.com
- Stanford Health Care: Osteogenesis Imperfecta (Abruf: 03.07.2019), stanfordhealthcare.org
- UpToDate, Inc.: Osteogenesis imperfecta: Management and prognosis (Abruf: 03.07.2019), uptodate.com
- Brittle Bone Society: What is OI? (Abruf: 03.07.2019), brittlebone.org
- GFMK GmbH & Co. KG: Osteogenesis imperfecta - Informationen für Betroffene und Angehörige (Abruf: 03.07.2019), osteogenesis-imperfecta.net
Wichtiger Hinweis:
Dieser Artikel enthält nur allgemeine Hinweise und darf nicht zur Selbstdiagnose oder -behandlung verwendet werden. Er kann einen Arztbesuch nicht ersetzen.






